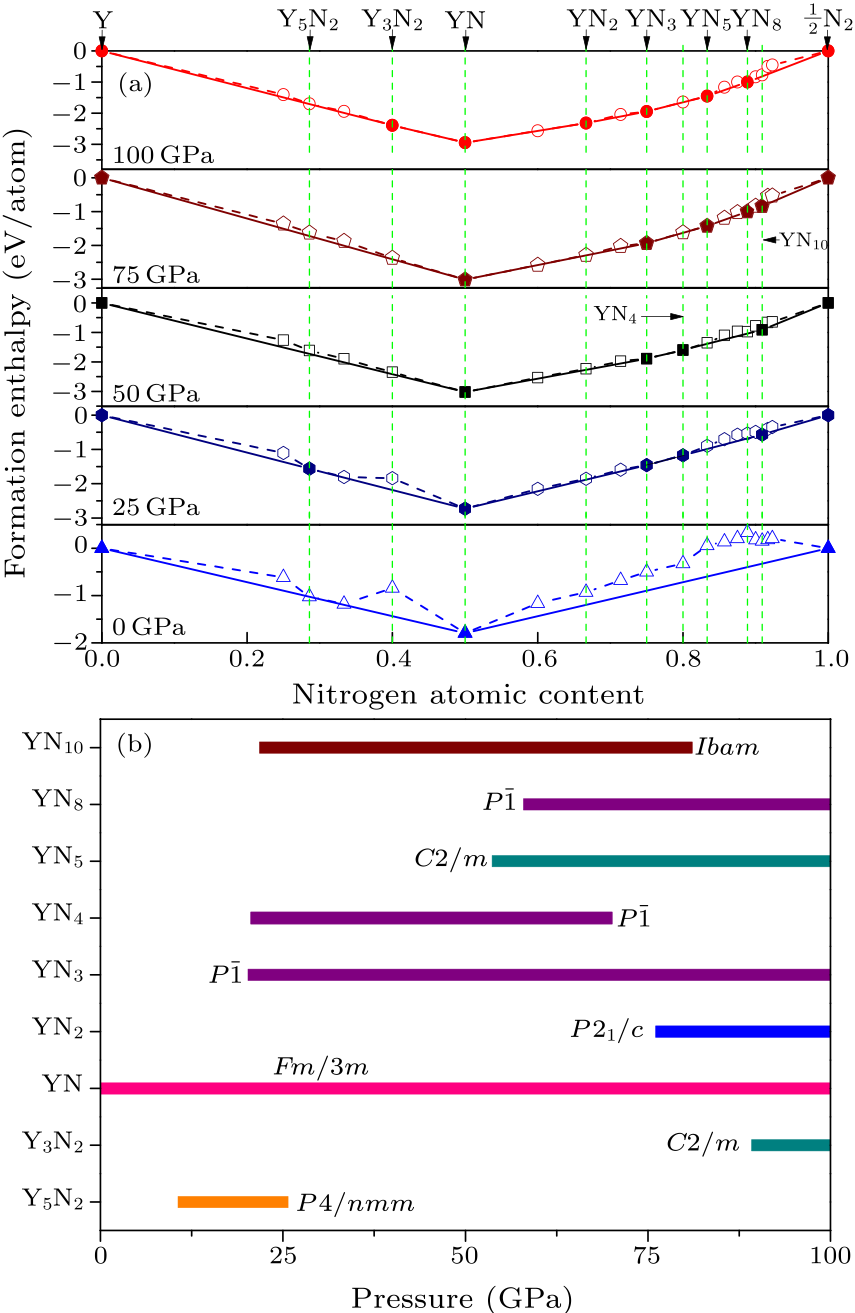
Fig. 1. (a) The convex hull of yttrium nitrides under different pressures. (b) The predicted stable pressure ranges of yttrium nitrides.
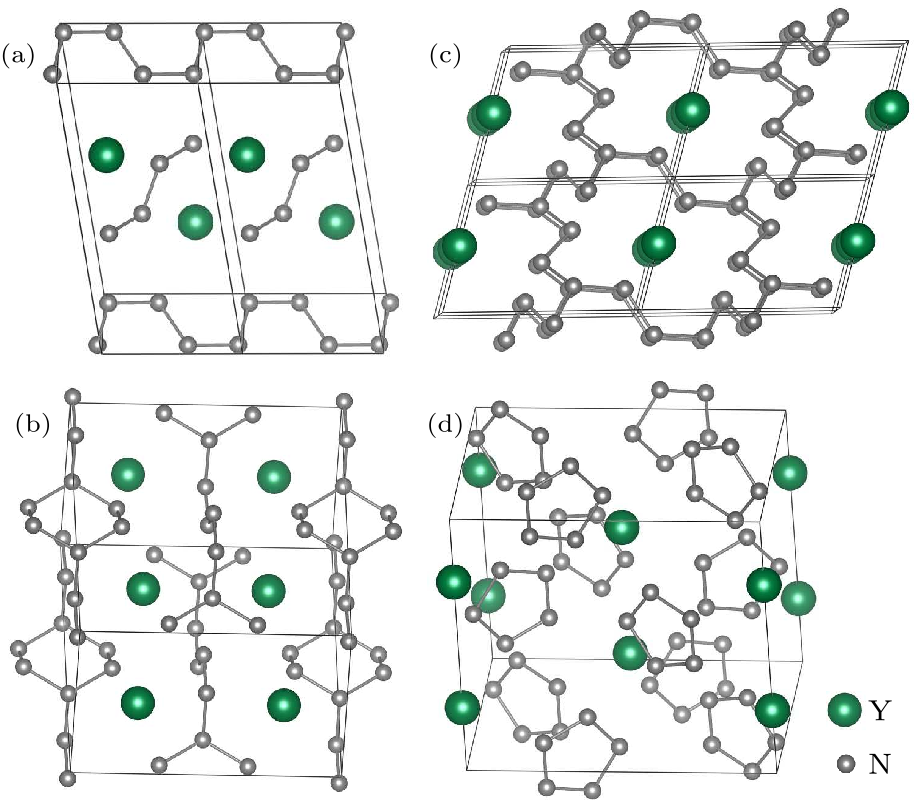
Fig. 2. Crystal structures of polynitrogens in yttrium nitrides. (a) The $P\bar{1}$ phase of YN$_{4}$ at 50 GPa. (b) The $C/2m$ structure of YN$_{5}$ at 100 GPa. (c) The polymeric $P\bar{1}$ phase at 75 GPa for YN$_{8}$. (d) Puckered plane N$_{5}$ ring $Ibam$ phase of YN$_{10}$ at 75 GPa.
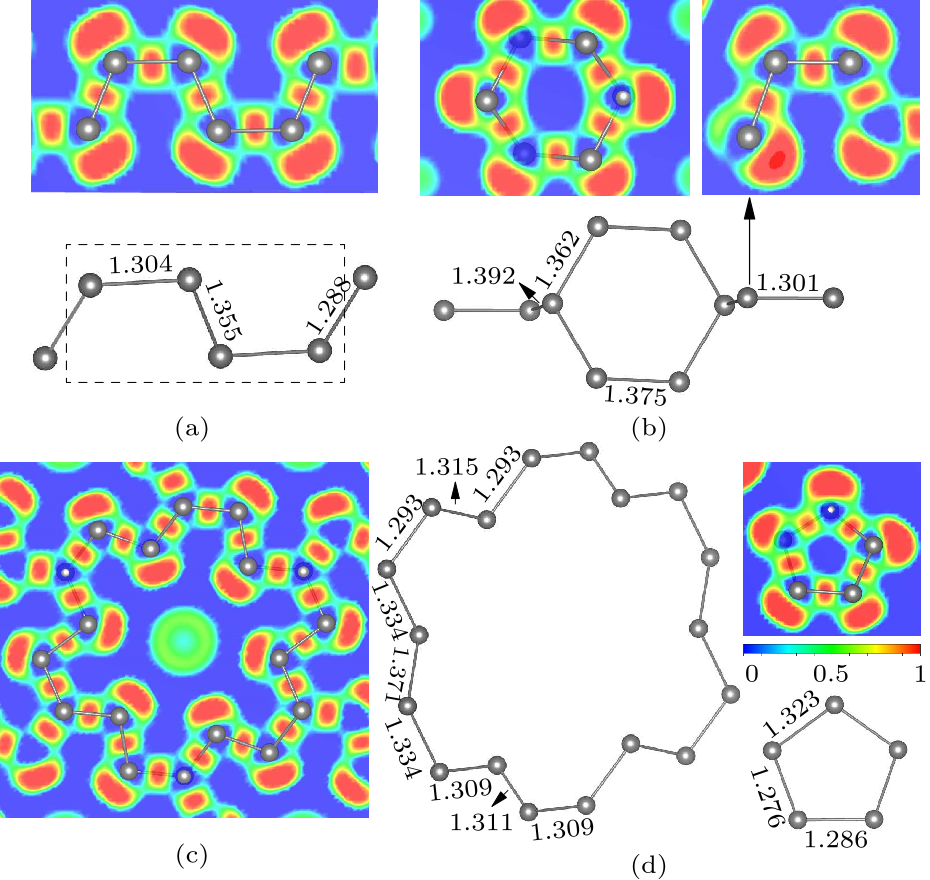
Fig. 3. ELFs and units of polynitrogens of yttrium nitrides. (a) The $P\bar{1}$ phase of YN$_{4}$ at 50 GPa. ELF plots in (0.34,0,1) sections for N$_{4}$ unit. (b) The $C/2m$ structure of YN$_{5}$ at 100 GPa. ELF plots in (0.65,0,$-$1) sections for N$_{10}$ unit. (c) The polymeric $P\bar{1}$ phase of YN$_{8}$ at 75 GPa. ELF plots in ($-$1,$-$1,1) sections for N$_{18}$ unit. (d) The puckered plane N$_{5}$ ring in $Ibam$ phase of YN$_{10}$ at 75 GPa. ELF plots in (0.48,1,0.3) sections for N$_{5}$ unit.
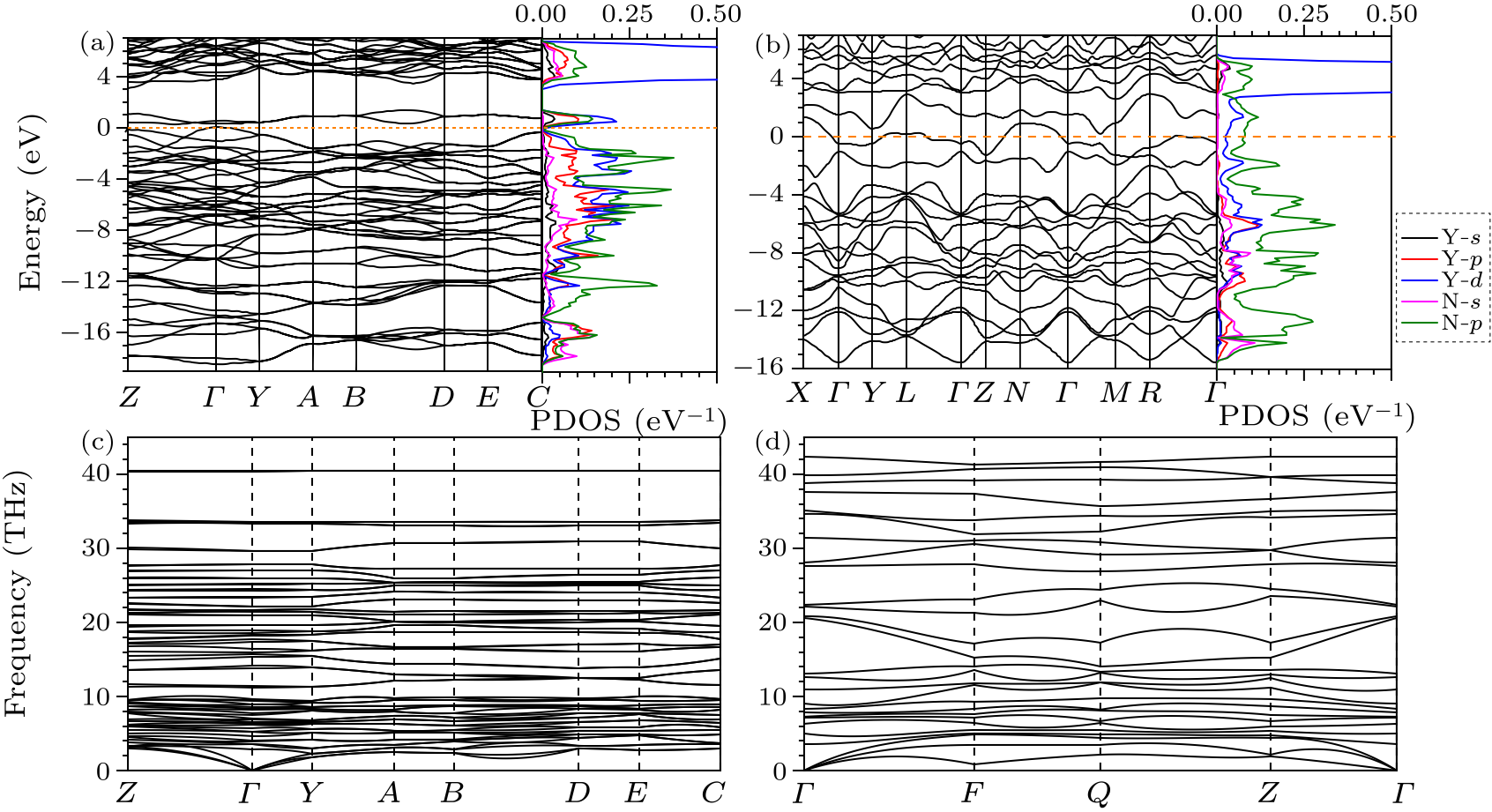
Fig. 4. Electronic band structure, DOS and phonon dispersion curves of yttrium nitrides. Band structures and DOS of $C2/m$ phase for YN$_{5}$ at 100 GPa (a) and $P\bar{1}$ phase of YN$_{8}$ at 75 GPa (b). Phonon dispersion curves of $C2/m$-YN$_{5}$ (c) and $P\bar{1}$-YN$_{8}$ (d) at ambient pressure.
| [1] | Christe K O, Wilson W W, Sheehy J A, and Boatz J A 1999 Angew. Chem. Int. Ed. 38 2004 | N5+: A Novel Homoleptic Polynitrogen Ion as a High Energy Density Material
| [2] | Zahariev F, Dudiy S, Hooper J, Zhang F et al. 2006 Phys. Rev. Lett. 97 155503 | Systematic Method to New Phases of Polymeric Nitrogen under High Pressure
| [3] | Eremets M I, Gavriliuk A G, Trojan I A, Dzivenko D A et al. 2004 Nat. Mater. 3 558 | Single-bonded cubic form of nitrogen
| [4] | Gregoryanz E, Goncharov A F, Sanloup C, and Metal S 2007 J. Chem. Phys. 126 184505 | High P-T transformations of nitrogen to 170GPa
| [5] | Jiang S, Holtgrewe N, Lobanov S S, Su F, Mahmood M F et al. 2018 Nat. Commun. 9 2624 | Metallization and molecular dissociation of dense fluid nitrogen
| [6] | Lei L, Tang Q, Zhang F, Liu S et al. 2020 Chin. Phys. Lett. 37 068101 | Evidence for a New Extended Solid of Nitrogen
| [7] | Peng F, Yao Y, Liu H, and Ma Y 2015 J. Phys. Chem. Lett. 6 2363 | Crystalline LiN 5 Predicted from First-Principles as a Possible High-Energy Material
| [8] | Peng F, Han Y, Liu H, and Yao Y 2015 Sci. Rep. 5 16902 | Exotic stable cesium polynitrides at high pressure
| [9] | Zhu S, Peng F, Liu H et al. 2016 Inorg. Chem. 55 7550 | Stable Calcium Nitrides at Ambient and High Pressures
| [10] | Shi X, Liu B, Yao Z, and Liu B 2020 Chin. Phys. Lett. 37 047101 | Pressure-Stabilized New Phase of CaN 4
| [11] | Ma S, Peng F, Zhu S, Li S et al. 2018 J. Phys. Chem. C 122 22660 | Novel Phase of AlN 4 as a Possible Superhard Material
| [12] | Wang X, Li J, Zhu H, Chen L et al. 2014 J. Chem. Phys. 141 044717 | Polymerization of nitrogen in cesium azide under modest pressure
| [13] | Zhang M, Yan H, Wei Q, Wang H et al. 2013 Europhys. Lett. 101 26004 | Novel high-pressure phase with pseudo-benzene “N 6 ” molecule of LiN 3
| [14] | Steele B A, Stavrou E, Crowhurst J C, Zaug J M et al. 2017 Chem. Mater. 29 735 | High-Pressure Synthesis of a Pentazolate Salt
| [15] | Peng F, Wang Y, Wang H, Zhang Y et al. 2015 Phys. Rev. B 92 094104 | Stable xenon nitride at high pressures
| [16] | Yin K, Wang Y, Liu H, Peng F et al. 2015 J. Mater. Chem. A 3 4188 | N 2 H: a novel polymeric hydronitrogen as a high energy density material
| [17] | Li Y, Feng X, Liu H et al. 2018 Nat. Commun. 9 722 | Route to high-energy density polymeric nitrogen t-N via He−N compounds
| [18] | Steele B A and Oleynik I I 2017 J. Phys. Chem. A 121 1808 | Pentazole and Ammonium Pentazolate: Crystalline Hydro-Nitrogens at High Pressure
| [19] | Raza Z, Pickard C, Pinilla C, and Saitta A 2013 Phys. Rev. Lett. 111 235501 | High Energy Density Mixed Polymeric Phase from Carbon Monoxide and Nitrogen
| [20] | Ashcroft N W 2004 Phys. Rev. Lett. 92 187002 | Hydrogen Dominant Metallic Alloys: High Temperature Superconductors?
| [21] | Prasad D L V K, Ashcroft N W, and Hoffmann R 2013 J. Phys. Chem. C 117 20838 | Evolving Structural Diversity and Metallicity in Compressed Lithium Azide
| [22] | Xia K, Zheng X, Yuan J, Liu C et al. 2019 J. Phys. Chem. C 123 10205 | Pressure-Stabilized High-Energy-Density Alkaline-Earth-Metal Pentazolate Salts
| [23] | Allred A L J 1961 J. Inorg. Nucl. Chem. 17 215 | Electronegativity values from thermochemical data
| [24] | Wang Y, Lv J, Zhu L, and Ma Y 2010 Phys. Rev. B 82 094116 | Crystal structure prediction via particle-swarm optimization
| [25] | Wang Y, Lv J, Zhu L, and Ma Y 2012 Comput. Phys. Commun. 183 2063 | CALYPSO: A method for crystal structure prediction
| [26] | Gao B, Gao P, Lu S, Lv J, Wang Y, and Ma Y 2019 Sci. Bull. 64 301 | Interface structure prediction via CALYPSO method
| [27] | Lv J, Wang Y, Zhu L, and Ma Y 2011 Phys. Rev. Lett. 106 015503 | Predicted Novel High-Pressure Phases of Lithium
| [28] | Zhu L, Wang H, Wang Y, Lv J et al. 2011 Phys. Rev. Lett. 106 145501 | Substitutional Alloy of Bi and Te at High Pressure
| [29] | Peng F, Miao M, Wang H, Li Q, and Ma Y 2012 J. Am. Chem. Soc. 134 18599 | Predicted Lithium–Boron Compounds under High Pressure
| [30] | Peng F, Sun Y, Pickard C J, Needs R J et al. 2017 Phys. Rev. Lett. 119 107001 | Hydrogen Clathrate Structures in Rare Earth Hydrides at High Pressures: Possible Route to Room-Temperature Superconductivity
| [31] | Peng F, Song X, Liu C, Li Q et al. 2020 Nat. Commun. 11 5227 | Xenon iron oxides predicted as potential Xe hosts in Earth’s lower mantle
| [32] | Lu C and Chen C 2020 Phys. Rev. Mater. 4 043402 | Indentation-strain stiffening in tungsten nitrides: Mechanisms and implications
| [33] | Lu C and Chen C 2020 Phys. Rev. Mater. 4 044002 | Structure-strength relations of distinct MoN phases from first-principles calculations
| [34] | Sun W, Kuang X, Keen H D J, Lu C et al. 2020 Phys. Rev. B 102 144524 | Second group of high-pressure high-temperature lanthanide polyhydride superconductors
| [35] | Chen B, Conway L J, Sun W, Kuang X et al. 2021 Phys. Rev. B 103 035131 | Phase stability and superconductivity of lead hydrides at high pressure
| [36] | Perdew J P and Wang Y 1992 Phys. Rev. B 45 13244 | Accurate and simple analytic representation of the electron-gas correlation energy
| [37] | Perdew J P, Burke K, and Ernzerhof M 1996 Phys. Rev. Lett. 77 3865 | Generalized Gradient Approximation Made Simple
| [38] | Kresse G and Furthmüller J 1996 Phys. Rev. B 54 11169 | Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set
| [39] | Blöchl P E 1994 Phys. Rev. B 50 17953 | Projector augmented-wave method
| [40] | Togo A, Oba F, and Tanaka I 2008 Phys. Rev. B 78 134106 | First-principles calculations of the ferroelastic transition between rutile-type and -type at high pressures
| [41] | Kempter C P, Krikorian N H, and McGuire J C 1957 Chem. Phys. 61 1237 | The Crystal Structure of Yttrium Nitride
| [42] | Bader R F W 1990 Atoms in Molecules: A Quantum Theory (Oxford: Clarendon) |
| [43] | Becke A D and Edgecombe K E 1990 J. Chem. Phys. 92 5397 | A simple measure of electron localization in atomic and molecular systems
| [44] | Jia Y, Zhao J, Zhang S, Yu S et al. 2019 Chin. Phys. Lett. 36 087401 | Superconductivity in Topological Semimetal θ -TaN at High Pressure *
| [45] | Wu Z, Zhao E, Xiang H, Hao X et al. 2007 Phys. Rev. B 76 054115 | Crystal structures and elastic properties of superhard and from first principles
| [46] | Evans W J, Lipp M J, Yoo C S, Cynn H et al. 2006 Chem. Mater. 18 2520 | Pressure-Induced Polymerization of Carbon Monoxide: Disproportionation and Synthesis of an Energetic Lactonic Polymer