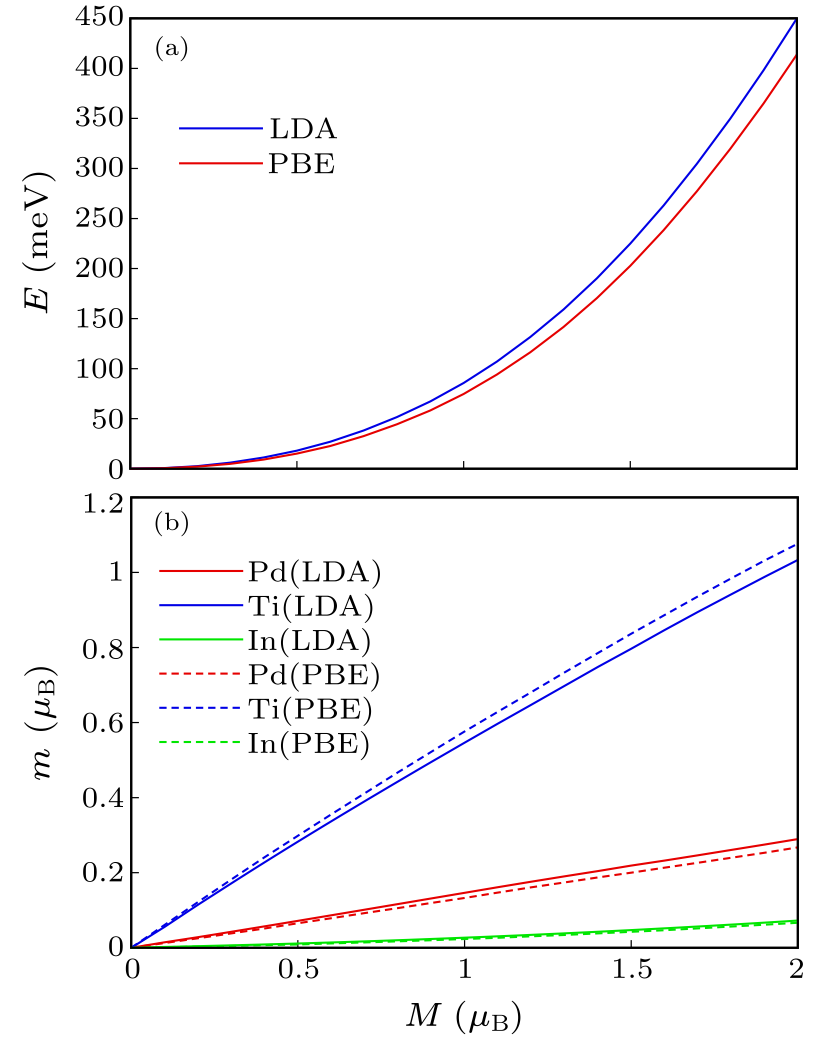
Fig. 1. Fixed spin moment energy as a function of magnetization on a per formula unit basis, using the LDA and PBE functionals (a), and moment in single LAPW spheres in the fixed spin moment calculations (b). The fixed spin moment calculations were performed in a scalar relativistic approximation, with LAPW sphere radii of 2.5 bohr.

Fig. 2. Band structure (a) and electronic density of states (DOS) and projections of $d$ character onto Pd and Ti, on a per formula unit basis (b). The Fermi level $E_{\rm F}$ is at 0 eV. These were calculated with the PBE functional including spin orbit. The projections were onto LAPW spheres of radius 2.5 bohr. (c) The DOS for bulk Pd metal. Note the position of the $d$-bands relative to $E_{\rm F}$.
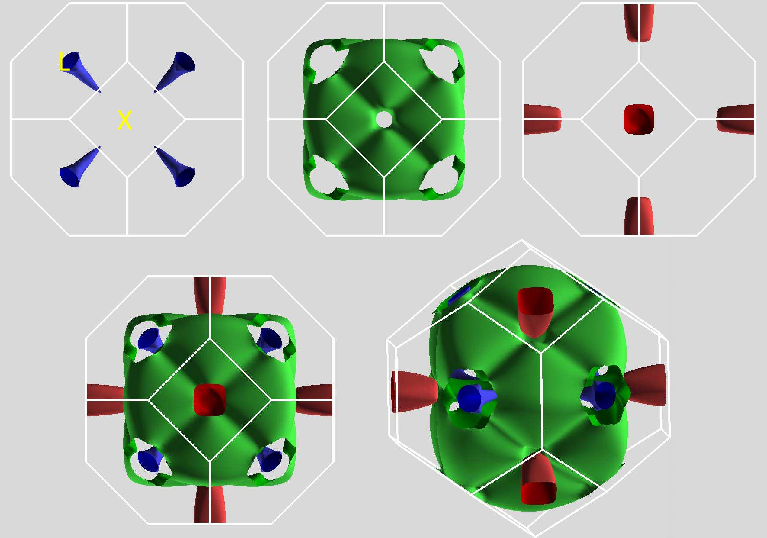
Fig. 3. Fermi surface as obtained with the PBE functional including SOC. The top panel shows the small anisotropic $L$ centered hole pockets (blue), the large main sheet (green) and the small $X$ electron pockets (red). The bottom shows two views of the Fermi surface with all three sheets.
| Configuration | Energy (eV) | M ($\mu_{\rm B}$/cell) | $m$ ($\mu_{\rm B}$) |
|---|---|---|---|
| ideal | 0 | 0.00 | 0.00 |
| Pd$_{\rm In}$ In$_{\rm Pd}$ | 1.21 | 0.00 | 0.00 |
| Ti$_{\rm In}$ In$_{\rm Ti}$ | 1.15 | 0.32 | 0.22 |
| Ti$_{\rm Pd}$ Pd$_{\rm Ti}$ | 1.08 | 1.39 | 0.67 |
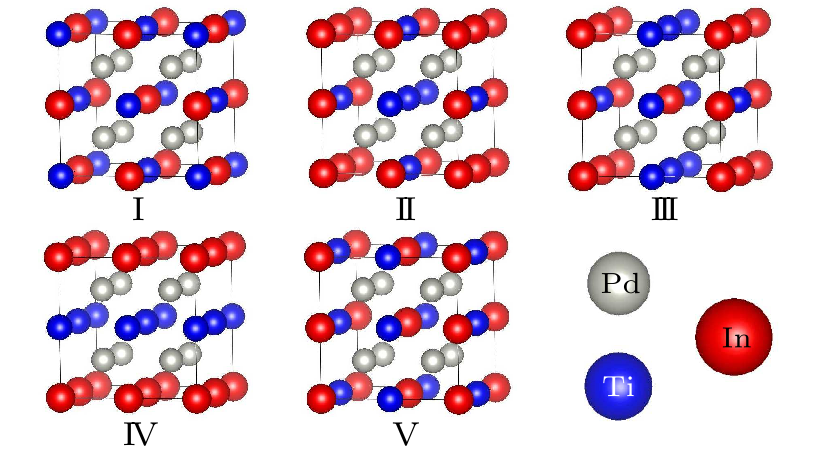
Fig. 4. Supercell structures for the ideal structure (I) and structures with 50% swap disorder between Ti and In. The gray, cyan and purple spheres represent Pd, Ti, and In, respectively.
| Config. | Energy (eV) | $M$ ($\mu_{\rm B}$) | $M_{\rm Pd}$ ($\mu_{\rm B}$) | $M_{\rm Ti}$ ($\mu_{\rm B}$) |
|---|---|---|---|---|
| I (ideal) | 0 | 0.00 | 0.00 | 0.00 |
| II | 0.24 | 0.00 | 0.00 | 0.00 |
| III | 1.15 | 0.64 | 0.06 | 0.48 |
| IV | 0.59 | 0.00 | 0.00 | 0.00 |
| V | 2.19 | 1.67 | 0.26 | 1.13 |
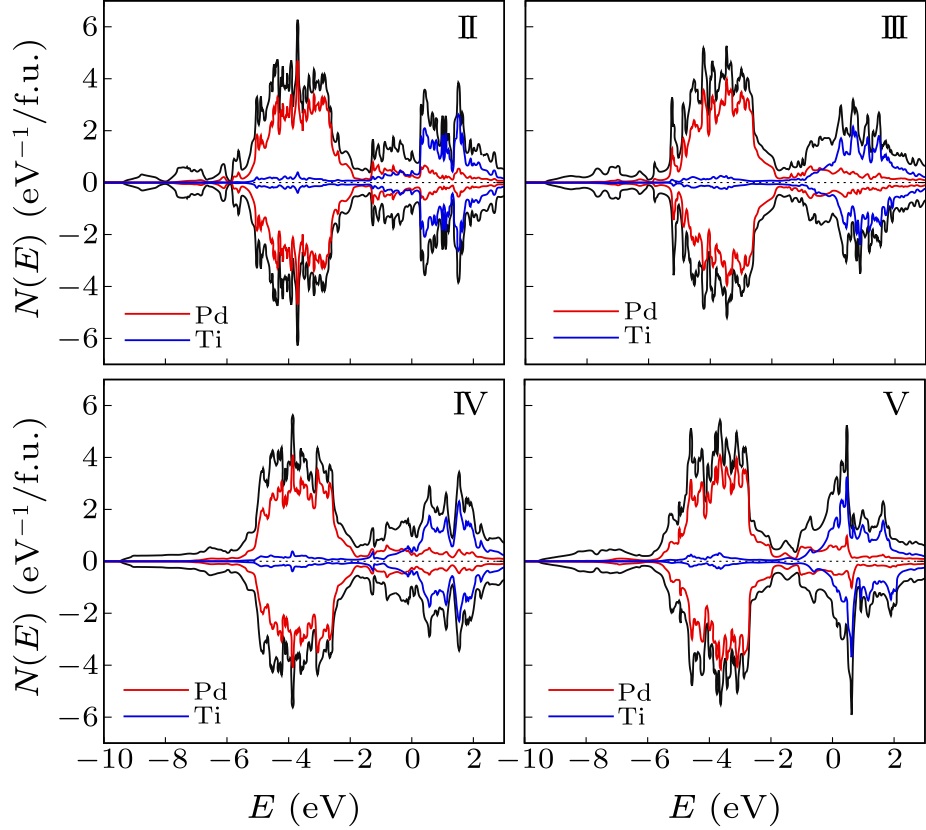
Fig. 5. Electronic density of states for the structures II, III, IV and V, as shown in Fig. 4. These are on a per formula unit basis with majority spin shown as positive (above the axis) and minority spin as negative. The Pd and Ti projections are onto atom centered spheres of radius 2.35 Bohr.
| Site | $x$ | $y$ | $z$ |
|---|---|---|---|
| Pd 8$g$ | 0.2312 | 0.2312 | 0.7688 |
| Ti1 3$d$ | 0.5 | 0.0 | 0.0 |
| Ti2 1$a$ | 0.0 | 0.0 | 0.0 |
| In1 1$b$ | 0.5 | 0.5 | 0.5 |
| In2 3$c$ | 0.0 | 0.5 | 0.5 |
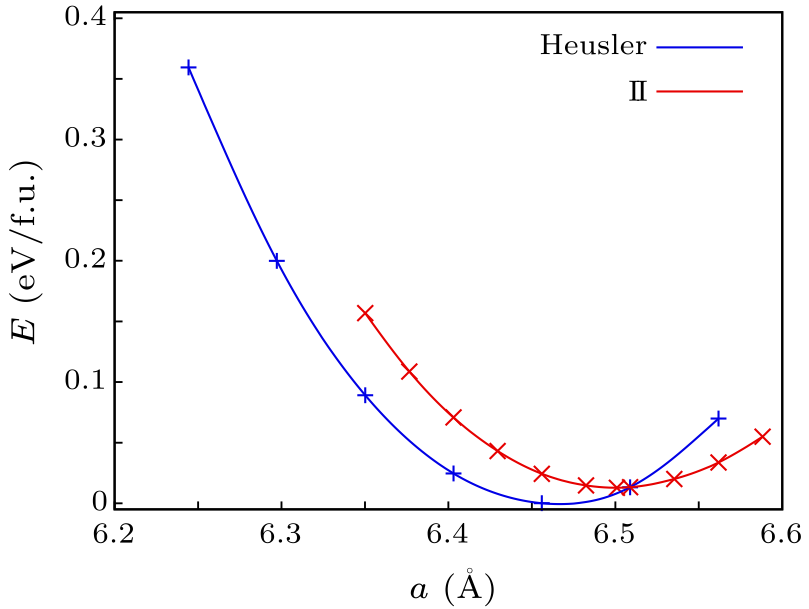
Fig. 6. Energy as a function of lattice parameter as obtained with the PBE functional for the full Heusler structure and the new structure, II. Energies are per formula unit, with the energy zero as the lowest energy for the full Heusler structure.
| [1] | Heusler F 1903 Verhandlungen Deutschen Physikalischen Ges. 5 219 |
| [2] | Graf T, Felser C and Parkin S S P 2011 Prog. Solid State Chem. 39 1 | Simple rules for the understanding of Heusler compounds
| [3] | Kandpal H C, Fecher G H and Felser C 2007 J. Phys. D 40 1507 | Calculated electronic and magnetic properties of the half-metallic, transition metal based Heusler compounds
| [4] | Palmstrom C J 2016 Prog. Cryst. Growth Charact. Mater. 62 371 | Heusler compounds and spintronics
| [5] | Jourdan M, Minar J, Braun J, Kronenberg A, Chadov S, Balke B, Glskovskii A, Kolbe M, Elmers H J, Schonhense G, Ebert H, Felser C and Klaui M 2014 Nat. Commun. 5 3974 | Direct observation of half-metallicity in the Heusler compound Co2MnSi
| [6] | Gersci Z, Rajanikanth A, Takahashi Y K, Hono K, Kikuchi M, Tezuka N and Inomata K 2006 Appl. Phys. Lett. 89 082512 | Spin polarization of Co2FeSi full-Heusler alloy and tunneling magnetoresistance of its magnetic tunneling junctions
| [7] | Neumann K U, Crangle J, Kremer R K, Zayer N K and Ziebeck K R A 1993 J. Magn. Magn. Mater. 127 47 | Magnetic order in Pd2TiIn: A new itinerant antiferromagnet?
| [8] | Kämmerer S, Thomas A, Hütten A and Reiss G 2004 Appl. Phys. Lett. 85 79 | Co2MnSi Heusler alloy as magnetic electrodes in magnetic tunnel junctions
| [9] | Balke B, Fecher G H, Kandpal H C, Felser C, Kobayashi K, Ikenaga E, Kim J J and Ueda S 2006 Phys. Rev. B 74 104405 | Properties of the quaternary half-metal-type Heusler alloy
| [10] | Ouardi S, Fecher G H, Felser C and Kübler J 2013 Phys. Rev. Lett. 110 100401 | Realization of Spin Gapless Semiconductors: The Heusler Compound
| [11] | Şaşıoğlu E, Sandratskii L M and Bruno P 2004 Phys. Rev. B 70 024427 | First-principles calculation of the intersublattice exchange interactions and Curie temperatures of the full Heusler alloys
| [12] | Ishida S, Kashiwagi S, Fujii S and Asano S 1995 Physica B 210 140 | Magnetic and half-metallic properties of new Heusler alloys Ru2MnZ (Z Si, Ge, Sn and Sb)
| [13] | Luo H, Liu G, Meng F, Wang L, Liu E, Wu G, Zhu X and Jiang C 2011 Comput. Mater. Sci. 50 3119 | Slater–Pauling behavior and half-metallicity in Heusler alloys Mn2CuZ (Z=Ge and Sb)
| [14] | Gofryk K, Kaczorowski D, Plackowski T, Leithe-Jasper A and Grin Y 2005 Phys. Rev. B 72 094409 | Magnetic and transport properties of the rare-earth-based Heusler phases and
| [15] | Qin G, Ren W and Singh D J 2020 Phys. Rev. B 101 014427 | Interplay of local moment and itinerant magnetism in cobalt-based Heusler ferromagnets: and
| [16] | Mondal C, Barman C K, Pathak B and Alam A 2019 Phys. Rev. B 100 245151 | Type-II Dirac states in full Heusler compounds ( = Ti, Zr, and Hf)
| [17] | Hinterleitner B, Knapp I, Poneder M, Shi Y, Muller H, Eguchi G, Eisenmenger-Sittner C, Stoger-Pollach M, Kakefuda Y, Kawamoto N, Guo Q, Baba T, Mori T, Ullah S, Chen X Q and Bauer E 2019 Nature 576 85 | Thermoelectric performance of a metastable thin-film Heusler alloy
| [18] | Tsujii N, Nishide A, Hayakawa J and Mori T 2019 Sci. Adv. 5 eaat5935 | Observation of enhanced thermopower due to spin fluctuation in weak itinerant ferromagnet
| [19] | Neumann K U, Crangle J, Smith J G and Ziebeck K R A 1994 J. Magn. Magn. Mater. 137 264 | Ferromagnetism in an intermetallic compound containing palladium, titanium and aluminium
| [20] | Szymanski K, Biernacka M, Dobrzynski L, Neumann K U, Perzynska K, Satula D, Zaleski P and Ziebeck K R A 1999 J. Magn. Magn. Mater. 195 651 | Pd2TiAl: investigated by monochromatic, circularly polarized Mössbauer source
| [21] | Jezierski A 1995 Phys. Stat. Sol. (b) 190 471 | Density of States in Ordered and Disordered Pd2TiAl and Pd2TiSn Heusler Alloys
| [22] | Neumann K U, Crangle J, Smith J G, Zayer N K and Ziebeck K R A 1995 J. Magn. Magn. Mater. 140 185 | Magnetic order in ternary intermetallic compounds containing palladium and titanium
| [23] | Liddiard A J, Brown D, Crapper M D, Petty M, Smith J G, Telling N D, Bedwell K H, Flannery L B and Skull P A 1996 J. Phys.: Condens. Matter 8 3955 | The experimental density of states of Pd 4d and Ti 3d in the intermetallic compound
| [24] | Jezierski A, Pugacheva M, Morkowski J A and Szajek A 1997 Acta Phys. Pol. A 91 151 | Electronic Structure and Magnetic Properties of Intermetallic Alloys
| [25] | Ouladdiaf B, Neumann K U, Crangle J, Zayer N K, Ziebeck K R A and Ressouche E 1994 J. Phys.: Condens. Matter 6 1563 | A neutron diffraction study of the phase transition in Pd 2 TiIn
| [26] | Jaswal S S 1995 J. Magn. Magn. Mater. 139 L1 | Is ThInPd2 magnetic?
| [27] | Jezierski A, Morkowski J A, Szajek A and Pugacheva M 1995 J. Phys.: Condens. Matter 7 4447 | Electronic structure in ternary intermetallic Pd 2 TiX (X=Al,Ga,In) Heusler-type alloys: are they magnetic?
| [28] | Zhang Q, Ren Z, Wu N, Wang W, Gao Y, Zhang Q, Shi J, Zhuang L, Sun X and Fu L 2018 npj 2D Mater. Appl. 2 22 | Bioactive Natural Products from Endophytic Microbes
| [29] | Lei B H, Fu Y, Feng Z and Singh D J 2020 Phys. Rev. B 101 020404(R) | Quantum critical point and ferromagnetic semiconducting behavior in -type
| [30] | Singh D J and Mazin I I 1998 Phys. Rev. B 57 14352 | Electronic structure, local moments, and transport in
| [31] | Feng Y, Rhee J Y, Wiener T A, Lynch D W, Hubbard B E, Sievers A J, Schlagel D L, Lograsso T A and Miller L L 2001 Phys. Rev. B 63 165109 | Physical properties of Heusler-like
| [32] | Bilc D I and Ghosez P 2011 Phys. Rev. B 83 205204 | Electronic and thermoelectric properties of Fe VAl: The role of defects and disorder
| [33] | Singh D J and Nordstrom L 2006 Planewaves Pseudopoten-tials and the LAPW Method 2nd edn (Berlin: Springer) |
| [34] | Blaha P, Schwarz K, Tran F, Laskowski R, Madsen G K H and Marks L D 2020 J. Chem. Phys. 152 074101 | WIEN2k: An APW+lo program for calculating the properties of solids
| [35] | Singh D 1991 Phys. Rev. B 43 6388 | Ground-state properties of lanthanum: Treatment of extended-core states
| [36] | Sjostedt E, Nordstrom L and Singh D J 2000 Solid State Commun. 114 15 | An alternative way of linearizing the augmented plane-wave method
| [37] | Perdew J P, Burke K and Ernzerhof M 1996 Phys. Rev. Lett. 77 3865 | Generalized Gradient Approximation Made Simple
| [38] | Madsen G K H and Singh D J 2006 Comput. Phys. Commun. 175 67 | BoltzTraP. A code for calculating band-structure dependent quantities
| [39] | Skaftouros S, Ozdogan K, Sasioglu E and Galanakis I 2013 Phys. Rev. B 87 024420 | Generalized Slater-Pauling rule for the inverse Heusler compounds
| [40] | Han Y, Chen Z, Kuang M, Liu Z, Wang X and Wang X 2019 Results Phys. 12 435 | 171 Scandium-based full Heusler compounds: A comprehensive study of competition between XA and L21 atomic ordering
| [41] | He J, Amsler M, Xia Y, Naghavi S S, Hegde V I, Hao S, Goedecker S, Ozolins V and Wolverton C 2016 Phys. Rev. Lett. 117 046602 | Ultralow Thermal Conductivity in Full Heusler Semiconductors
| [42] | Gurth M, Rogl G, Romaka V V, Grytsiv A, Bauer E and Rogl P 2016 Acta Mater. 104 210 | Thermoelectric high ZT half-Heusler alloys Ti1−x−yZrxHfyNiSn (0 ≤ x ≤ 1; 0 ≤ y ≤ 1)
| [43] | Zhu H, Mao J, Li Y, Sun J, Wang Y, Zhu Q, Song Q, Zhou J, Fu Y, He R, Tong T, Liu Z, Ren W, Wang Z, Luo J, Sotnikov A, Bao J, Nielsch K, Chen G, Singh D J and Ren Z 2019 Nat. Commun. 10 270 | Discovery of TaFeSb-based half-Heuslers with high thermoelectric performance
| [44] | Rausch E, Balke B, Deschauer T, Ouradi S and Felser C 2015 APL Mater. 3 041516 | Charge carrier concentration optimization of thermoelectric p -type half-Heusler compounds
| [45] | Feng Z, Fu Y, Zhang Y and Singh D J 2020 Phys. Rev. B 101 064301 | Characterization of rattling in relation to thermal conductivity: Ordered half-Heusler semiconductors
| [46] | Gornicka K, Kuderowicz G, Carnicom E M, Kutorasinski K, Wiendlocha B, Cava R J and Klimczuk T 2020 Phys. Rev. B 102 024507 | Soft-mode enhanced type-I superconductivity in
| [47] | Pauling L 1947 J. Am. Chem. Soc. 69 542 | Atomic Radii and Interatomic Distances in Metals
| [48] | Schwarz K and Mohn P 1984 J. Phys. F 14 L129 | Itinerant metamagnetism in YCO 2
| [49] | Lechermann F, Welsch F, Elsasser C, Ederer C, Fahnle M, Sanchez J M and Meyer B 2002 Phys. Rev. B 65 132104 | Density-functional study of LSDA versus GGA
| [50] | Fu Y and Singh D J 2019 Phys. Rev. B 100 045126 | Density functional methods for the magnetism of transition metals: SCAN in relation to other functionals
| [51] | Moriya T 1985 Spin Fluctuations in Itinerant Electron Magnetism (Berlin: Springer) |
| [52] | Larson P, Mazin I I and Singh D J 2004 Phys. Rev. B 69 064429 | Magnetism, critical fluctuations, and susceptibility renormalization in Pd
| [53] | Putatunda A, Qin G, Ren W and Singh D J 2020 Phys. Rev. B 102 014442 | Competing magnetic orders in quantum critical
| [54] | Dudarev S L, Botton G A, Savrasov S Y, Humphreys C J and Sutton A P 1998 Phys. Rev. B 57 1505 | Electron-energy-loss spectra and the structural stability of nickel oxide:??An LSDA+U study
| [55] | Cococcioni M and de Gironcoli S 2005 Phys. Rev. B 71 035105 | Linear response approach to the calculation of the effective interaction parameters in the method
| [56] | Tran F, Koller D and Blaha P 2012 Phys. Rev. B 86 134406 | Application of screened hybrid functionals to the bulk transition metals Rh, Pd, and Pt
| [57] | Brown D, Crapper M D, Bedwell K H, Petty M, Smith J G and Telling N D 1997 J. Phys.: Condens. Matter 9 2955 | A photoemission investigation of the valence band electronic structure of and
| [58] | Janak J F 1977 Phys. Rev. B 16 255 | Uniform susceptibilities of metallic elements
| [59] | Krasko G L 1987 Phys. Rev. B 36 8565 | Metamagnetic behavior of fcc iron
| [60] | Andersen O K, Madsen J, Poulsen U K, Jepsen O and Kollar J 1977 Physica B+C 86 249 | Magnetic ground state properties of transition metals
| [61] | Mazin I I and Singh D J 1997 Phys. Rev. B 56 2556 | Electronic structure and magnetism in Ru-based perovskites
| [62] | McGuire M A, Christianson A D, Sefat A S, Sales B C, Lumsden M D, Jin R, Payzant E A, Mandrus D, Luan Y, Keppens V, Varadarajan V, Brill J W, Hermann R P, Sougrati M T, Grandjean F and Long G J 2008 Phys. Rev. B 78 094517 | Phase transitions in LaFeAsO: Structural, magnetic, elastic, and transport properties, heat capacity and Mössbauer spectra
| [63] | Putatunda A and Singh D J 2019 Mater. Today Phys. 8 49 | Lorenz number in relation to estimates based on the Seebeck coefficient
| [64] | Jezierski A 2000 J. Phys.: Condens. Matter 12 8127 | Effect of vacancies on the electronic structure of Pd x TiSn alloys