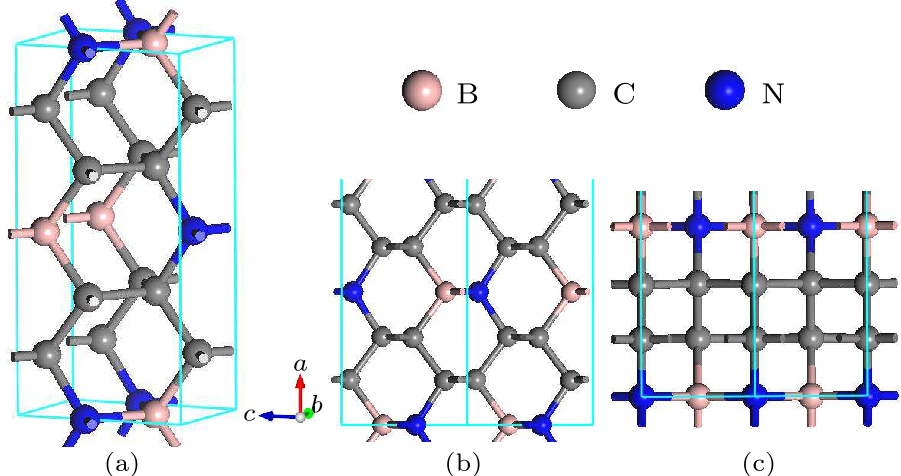
Fig. 1. Structures of $o$-BC$_{4}$N: (a) three-dimensional topology, (b) along the [010] direction, and (c) along the [001] direction.
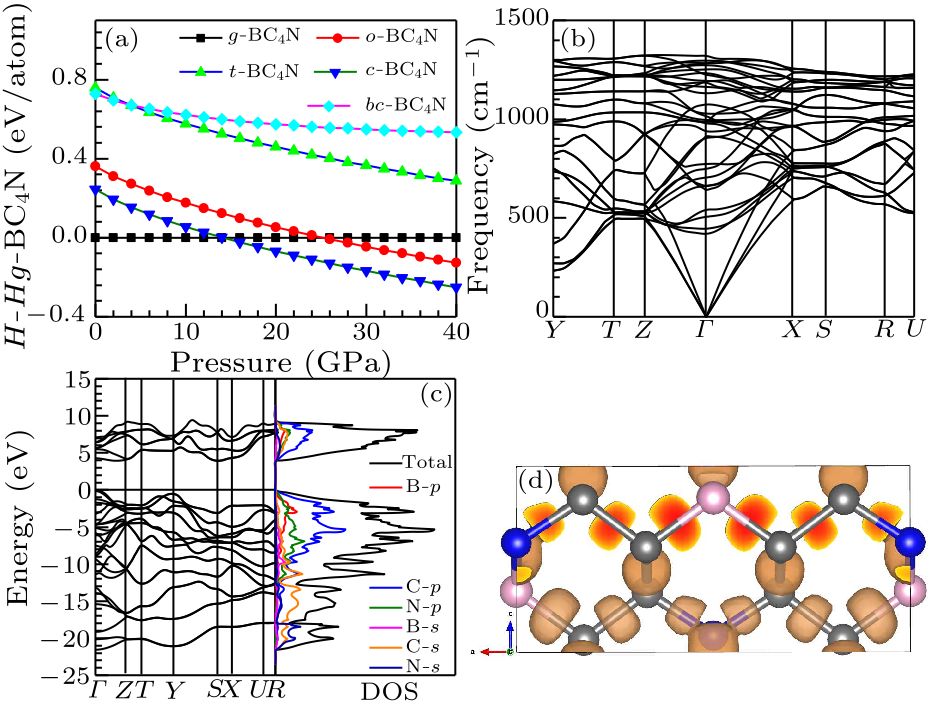
Fig. 2. (a) Enthalpy as a function of pressure for BC$_{4}$N allotropes relative to $g$-BC$_{4}$N. (b) The phonon-dispersion curves of $o$-BC$_{4}$N at a pressure of 0 GPa. (c) The band structure (left panel) and density of states (DOS) (right panel) of $o$-BC$_{4}$N. Zero denotes the energy measured from the valence top. (d) The conventional unit cell of $o$-BC$_{4}$N with ELF isosurface with an ELF value of 0.75.
| Phase | $o$-BC$_{4}$N | $t$-BC$_{4}$N | $c$-BC$_{4}$N | ||
|---|---|---|---|---|---|
| This work | This work | Ref. [ | This work | Ref. [ |
|
| Symmetry | $Imm2$ | $I\bar{4}m2$ | $I\bar{4}m2$ | $P3m1$ | $P3m1$ |
| $a$ | 7.651 | 2.552 | 2.547 | 2.538 | 2.507 |
| $b$ | 2.539 | 2.552 | 2.547 | 2.538 | 2.507 |
| $c$ | 3.614 | 10.964 | 10.947 | 6.267 | 6.196 |
| $\rho$ | 3.447 | 3.389 | 3.407 | 3.461 | 3.588 |
| $C_{11}$ | 1027.2 | 930.4 | 965.6 | 1056.0 | 1131.5 |
| $C_{22}$ | 1038.9 | ||||
| $C_{33}$ | 933.2 | 854.6 | 872.0 | 1083.2 | 1116.9 |
| $C_{44}$ | 498.0 | 303.3 | 381.8 | 425.2 | 508.5 |
| $C_{55}$ | 502.3 | ||||
| $C_{66}$ | 391.8 | 376.0 | 335.4 | 477.5 | 527.9 |
| $C_{12}$ | 24.4 | 39.1 | 40.8 | 101.0 | 75.8 |
| $C_{13}$ | 125.6 | 130.6 | 141.3 | 63.2 | 59.5 |
| $C_{23}$ | 133.7 | ||||
| $C_{15}$ | 47.9 | $-$6.7 | |||
| $B_{V}$ | 396.30 | 368.44 | 383.36 | 405.57 | 418.83 |
| $B_{R}$ | 396.28 | 368.41 | 383.35 | 405.56 | 418.77 |
| $G_{V}$ | 459.4 | 357.5 | 385.1 | 463.5 | 521.3 |
| $G_{R}$ | 452.6 | 350.1 | 380.9 | 461.0 | 521.1 |
| $B$ | 396.3 | 368.4 | 383.4 | 405.6 | 418.8 |
| $G$ | 456.0 | 353.8 | 383.0 | 462.2 | 521.2 |
| $E$ | 988.8 | 804.1 | 861.9 | 1004.9 | 1105.1 |
| $B/G$ | 0.87 | 1.04 | 1.00 | 0.88 | 0.80 |
| $\upsilon$ | 0.08 | 0.14 | 0.13 | 0.09 | 0.06 |
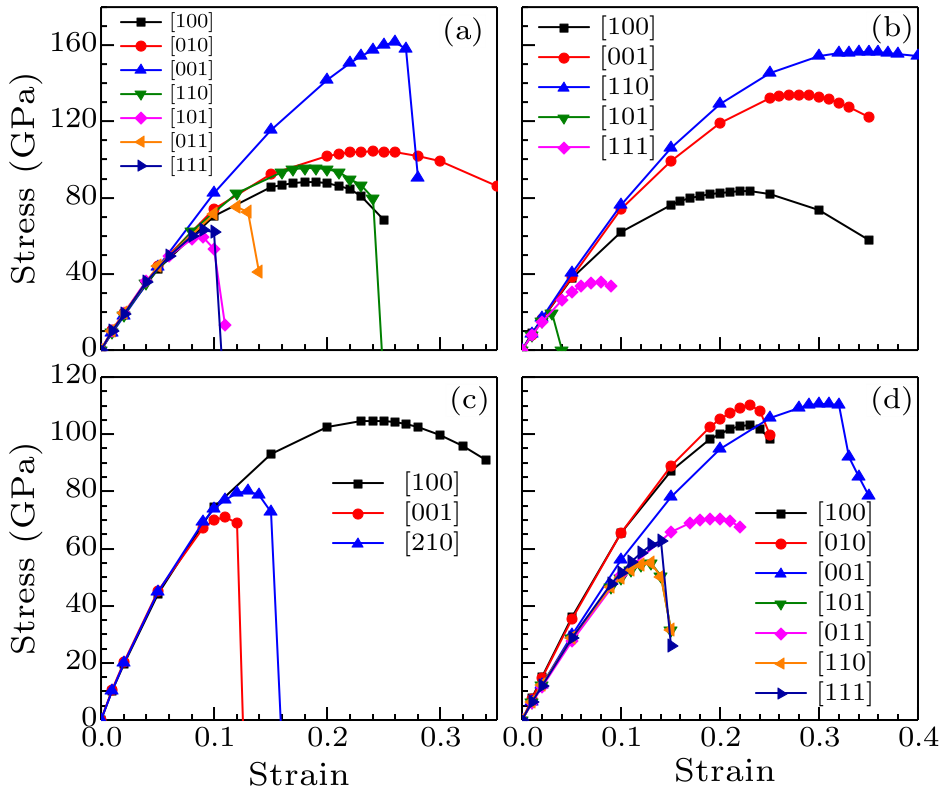
Fig. 3. Calculated tensile stress versus tensile strain of $o$-BC$_{4}$N(a), t-BC$_{4}$N(b), $c$-BC$_{4}$N(c)and $bc$-BC$_{4}$N(d) in principal symmetry directions.
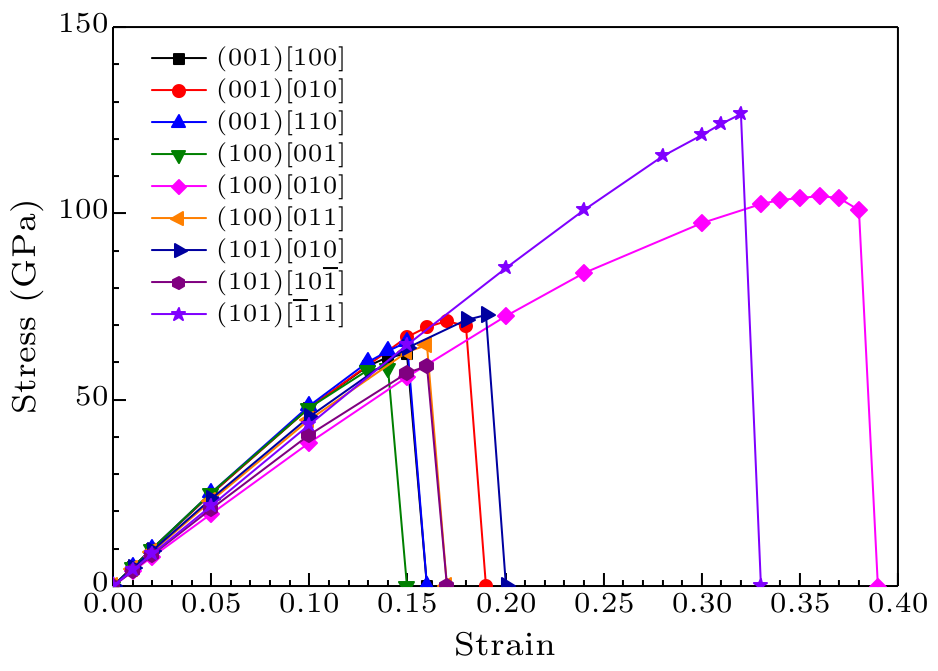
Fig. 4. Calculated shear stress versus shear strain for $o$-BC$_{4}$N in principal symmetry directions.
| Phase | Bond | $n$ | $d^{\mu}$ (Å) | $v_{{\rm b}}^{\mu}$ (Å$^{3}$) | $N_{\rm e}^{\mu}$ (Å$^{-3}$) | $E_{\rm h}^{\mu}$ (eV) | $E_{\rm g}^{\mu}$ (eV) | $f_{\rm i}^{\mu}$ | $H_{\rm v}^{\mu}$ (GPa) | $H_{\rm vav}$ (GPa) | $H_{\rm v}$ (Chen) (GPa) | $H_{\rm exp}$ (GPa) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| $o$-BC$_{4}$N | $C_{2}$-N | 4 | 1.535 | 2.782 | 0.652 | 13.603 | 16.421 | 0.314 | 62.1 | 78.7 | 81.7 | |
| C$_{1}$-C$_{2}$ | 4 | 1.539 | 2.803 | 0.882 | 13.514 | 13.528 | 0.002 | 109.3 | ||||
| BN | 4 | 1.579 | 3.025 | 0.662 | 12.684 | 15.025 | 0.287 | 60.3 | ||||
| B–C$_{1}$ | 4 | 1.618 | 3.252 | 0.455 | 11.942 | 12.031 | 0.015 | 61.2 | ||||
| C$_{1}$-C$_{2}$ | 8 | 1.547 | 2.844 | 0.756 | 13.352 | 13.364 | 0.002 | 97.5 | ||||
| $t$-BC$_{4}$N | C$_{1}$-N | 8 | 1.588 | 3.070 | 0.596 | 12.513 | 15.699 | 0.365 | 50.7 | 73.8 | 56.1 | |
| B–C$_{2}$ | 8 | 1.605 | 3.169 | 0.445 | 12.184 | 12.261 | 0.013 | 61.7 | ||||
| C$_{1}$-C$_{2}$ | 8 | 1.518 | 2.686 | 1.092 | 13.987 | 14.092 | 0.015 | 128.5 | ||||
| $c$-BC$_{4}$N | B–C$_{3}$ | 1 | 1.617 | 3.235 | 0.342 | 11.946 | 12.024 | 0.013 | 50.7 | 80.3 | 81.4 | 85$\pm$4$^{\rm a}$ |
| B-N | 3 | 1.566 | 2.938 | 0.735 | 12.944 | 15.031 | 0.258 | 68.3 | ||||
| C$_{4}$-C$_{3}$ | 3 | 1.550 | 2.850 | 0.843 | 13.278 | 13.278 | 0.000 | 104.5 | ||||
| C$_{4}$-C$_{1}$ | 1 | 1.554 | 2.872 | 0.349 | 13.193 | 13.193 | 0.000 | 57.7 | ||||
| C$_{2}$-C$_{1}$ | 3 | 1.549 | 2.850 | 0.849 | 13.306 | 13.307 | 0.000 | 105.2 | ||||
| C$_{2}$-N | 1 | 1.516 | 2.842 | 0.494 | 14.052 | 16.080 | 0.236 | 58.4 | ||||
| $bc$-BC$_{4}$N | $C_{4}$-N | 4 | 1.520 | 3.348 | 0.584 | 13.960 | 14.193 | 0.033 | 82.7 | 67.7 | 48.7 | |
| C$_{1}$-C$_{2}$ | 4 | 1.533 | 3.439 | 0.767 | 13.650 | 16.781 | 0.338 | 67.4 | ||||
| C$_{2}$-C$_{3}$ | 4 | 1.559 | 3.612 | 0.549 | 13.104 | 13.113 | 0.001 | 77.3 | ||||
| C$_{3}$-C$_{4}$ | 4 | 1.563 | 3.642 | 0.584 | 13.014 | 13.064 | 0.008 | 79.5 | ||||
| B-N | 4 | 1.596 | 3.612 | 0.249 | 12.358 | 12.385 | 0.004 | 42.9 | ||||
| B–C$_{1}$ | 4 | 1.600 | 3.875 | 0.734 | 12.280 | 14.209 | 0.253 | 65.2 | ||||
| $\beta$-BC$_{2}$N | C$_{1}$-C$_{2}$ | 4 | 1.528 | 2.744 | 0.911 | 13.767 | 13.767 | 0 | 114.1 | 75.5 | 76.9$^{\rm b}$ | 76$^{\rm c}$ |
| B–C$_{1}$ | 4 | 1.591 | 3.094 | 0.485 | 12.453 | 12.453 | 0 | 67.8 | ||||
| C$_{2}$-N | 4 | 1.578 | 3.021 | 0.662 | 12.706 | 14.49 | 0.231 | 64.6 | ||||
| B-N | 4 | 1.576 | 3.011 | 0.664 | 12.741 | 14.528 | 0.231 | 65.0 | ||||
| Diamond | C–C | 16 | 1.544 | 2.834 | 0.706 | 13.413 | 13.413 | 0.000 | 93.8 | 93.8 | 95.7$^{\rm b}$ | 96$\pm$5$^{\rm c}$ |
| $c$-BN | B-N | 16 | 1.569 | 2.973 | 0.673 | 12.889 | 14.691 | 0.230 | 66.4 | 66.4 | 65.2$^{\rm b}$ | 63$\pm$5$^{\rm c}$ |
|
Here $^{\rm a}$Ref. [ | ||||||||||||
| [1] | Haines J, Leger J and Bocquillon G 2001 Annu. Rev. Mater. Res. 31 1 | Synthesis and Design of Superhard Materials
| [2] | Liu A Y and Cohen M L 1989 Science 245 841 | Prediction of New Low Compressibility Solids
| [3] | Hanae N and Satoshi I 1996 J. Phys. Chem. Solids 57 41 | Structural stability of BC2N
| [4] | Solozhenko V L, Andrault D, Fiquet G, Mezouar M and Rubie D C 2001 Appl. Phys. Lett. 78 1385 | Synthesis of superhard cubic BC2N
| [5] | Zhao Y, He D W, Daemen L L, Shen T D, Schwarz R B, Zhu Y, Bish D L, Huang J, Zhang J, Shen G, Qian J and Zerda T W 2002 J. Mater. Res. 17 3139 | Superhard B–C–N materials synthesized in nanostructured bulks
| [6] | Tang M, He D, Wang W, Wang H, Xu C, Li F and Guan J 2012 Scr. Mater. 66 781 | Superhard solid solutions of diamond and cubic boron nitride
| [7] | Luo X G, Zhou X F, Liu Z Y, He J L, Xu B, Yu D L, Wang H T and Tian Y J 2008 J. Phys. Chem. C 112 9516 | Refined Crystal Structure and Mechanical Properties of Superhard BC 4 N Crystal: First-Principles Calculations
| [8] | Zhang Y, Sun H and Chen C F 2008 Phys. Rev. B 77 094120 | Influence of carbon content on the strength of cubic : A first-principles study
| [9] | Li F, Man Y H, Li C M, Wang J P and Chen Z Q 2015 Comput. Mater. Sci. 102 327 | Mechanical properties, minimum thermal conductivity, and anisotropy in bc-structure superhard materials
| [10] | Wang D, Shi R and Gan L H 2017 Chem. Phys. Lett. 669 80 | t-C8B2N2: A potential superhard material
| [11] | Guo W F, Wang L S, Li Z P, Xia M R and Gao F M 2015 Chin. Phys. Lett. 32 096201 | Urtra-Hard Bonds in P -Carbon Stronger than Diamond
| [12] | Gou H, Hou L, Zhang J, Sun G, Gao L and Gao F 2006 Appl. Phys. Lett. 89 141910 | Theoretical hardness of PtN2 with pyrite structure
| [13] | Wang Y, Lv J, Zhu L and Ma Y 2010 Phys. Rev. B 82 094116 | Crystal structure prediction via particle-swarm optimization
| [14] | Wang Y, Lv J, Zhu L and Ma Y 2012 Comput. Phys. Commun. 183 2063 | CALYPSO: A method for crystal structure prediction
| [15] | Li Q, Zhou D, Zheng W, Ma Y and Chen C 2013 Phys. Rev. Lett. 110 136403 | Global Structural Optimization of Tungsten Borides
| [16] | Li Y W, Hao J, Liu H Y, Lu S Y and Tse J S 2015 Phys. Rev. Lett. 115 105502 | High-Energy Density and Superhard Nitrogen-Rich B-N Compounds
| [17] | Hohenberg P and Kohn W 1964 Phys. Rev. 136 B864 | Inhomogeneous Electron Gas
| [18] | Kohn W and Sham L J 1965 Phys. Rev. 140 A1133 | Self-Consistent Equations Including Exchange and Correlation Effects
| [19] | Blöchl P E 1994 Phys. Rev. B 50 17953 | Projector augmented-wave method
| [20] | Perdew J P, Burke K and Ernzerhof M 1996 Phys. Rev. Lett. 77 3865 | Generalized Gradient Approximation Made Simple
| [21] | Ding Y C 2012 Physica B 407 2282 | Mechanical properties and hardness of new carbon-rich superhard C11N4 from first-principles investigations
| [22] | Baroni S, de Gironcoli S, dal Corso A and Giannozzi P 2001 Rev. Mod. Phys. 73 515 | Phonons and related crystal properties from density-functional perturbation theory
| [23] | Lazzeri M and Mauri F 2003 Phys. Rev. Lett. 90 036401 | First-Principles Calculation of Vibrational Raman Spectra in Large Systems: Signature of Small Rings in Crystalline
| [24] | Troullier N and Martins J L 1991 Phys. Rev. B 43 1993 | Efficient pseudopotentials for plane-wave calculations
| [25] | Roundy D, Krenn C R, Cohen M L and Morris J W Jr 1999 Phys. Rev. Lett. 82 2713 | Ideal Shear Strengths of fcc Aluminum and Copper
| [26] | Zhang R F, Veprek S and Argon A S 2008 Phys. Rev. B 77 172103 | Anisotropic ideal strengths and chemical bonding of wurtzite BN in comparison to zincblende BN
| [27] | Yang J, Sun H and Chen C 2008 J. Am. Chem. Soc. 130 7200 | Is Osmium Diboride An Ultra-Hard Material?
| [28] | Chen X Q, Fu C L and Podloucky R 2008 Phys. Rev. B 77 064103 | Bonding and strength of solid nitrogen in the cubic gauche (cg-N) structure
| [29] | Gao F, He J, Wu E, Liu S, Yu D, Li D, Zhang S and Tian Y 2003 Phys. Rev. Lett. 91 015502 | Hardness of Covalent Crystals
| [30] | Gao F, Xu R and Liu K 2005 Phys. Rev. B 71 052103 | Origin of hardness in nitride spinel materials
| [31] | Gao F 2006 Phys. Rev. B 73 132104 | Theoretical model of intrinsic hardness
| [32] | Cracknell A P, Davies B L, Miller S C and Love W F 1979 Kronecker Product Tables (New York: Plenum Press) |
| [33] | Savin A, Nesper R, Wengert S and Fässler T F 1997 Angew. Chem. Int. Ed. English 36 1808 | ELF: The Electron Localization Function
| [34] | Gutiérrez G, Menéndez-Proupin E and Singh A K 2006 J. Appl. Phys. 99 103504 | Elastic properties of the bcc structure of bismuth at high pressure
| [35] | Mouhat F and Coudert F X 2014 Phys. Rev. B 90 224104 |
| [36] | Hill R 1952 Proc. Phys. Soc. Sect. A 65 349 | The Elastic Behaviour of a Crystalline Aggregate
| [37] | Pugh S F 1954 Philos. Mag. A 45 823 | XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals
| [38] | Li Z, Gao F and Xu Z 2012 Phys. Rev. B 85 144115 | Strength, hardness, and lattice vibrations of -carbon and -carbon: First-principles calculations
| [39] | Li Z and Gao F 2012 Phys. Chem. Chem. Phys. 14 869 | Structure, bonding, vibration and ideal strength of primitive-centered tetragonal boron nitride
| [40] | Chen X Q, Niu H, Li D and Li Y 2011 Intermetallics 19 1275 | Modeling hardness of polycrystalline materials and bulk metallic glasses
| [41] | Chen X Q, Niu H, Franchini C, Li D Z and Li Y Y 2011 Phys. Rev. B 84 121405 | Hardness of -carbon: Density functional theory calculations