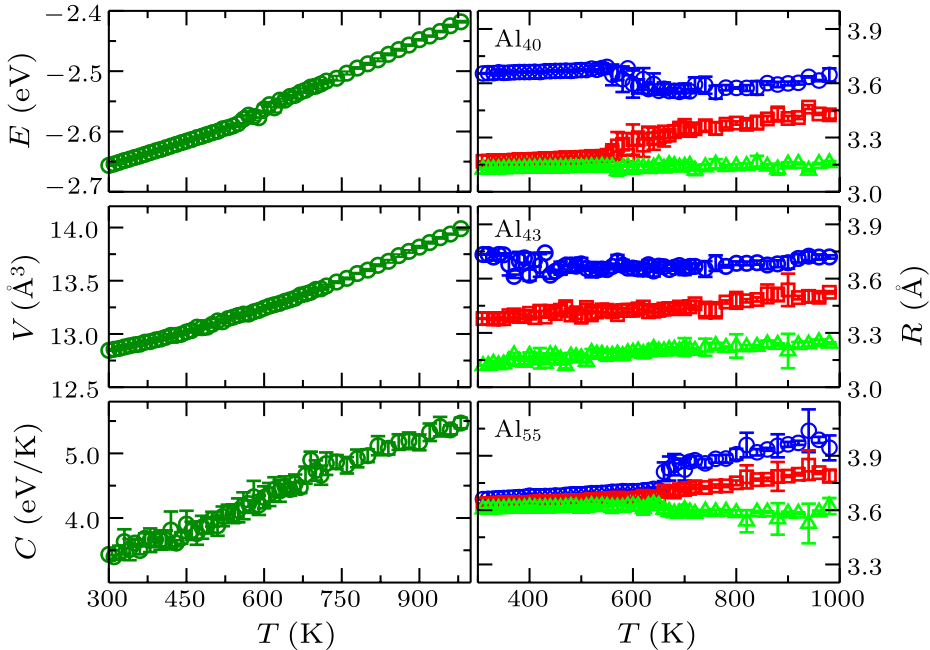
Fig. 1. (Color online) Left panel: the energy (upper), volume (middle) and specific heat (lower) as a function of temperature for Al$_{43}$. The continuous change shows a typical glass transition behavior. Right panel: three principal radii of gyration ($R_1,2,3$ indicated by different colors) for Al$_{40}$ (upper), Al$_{43}$ (middle), and Al$_{55}$ (lower). Around $T_{\rm s}$ ($\sim$350 K), the shape of Al$_{43}$ begins to change, while the shape of Al$_{40}$ and Al$_{55}$ is unchanged until melting.
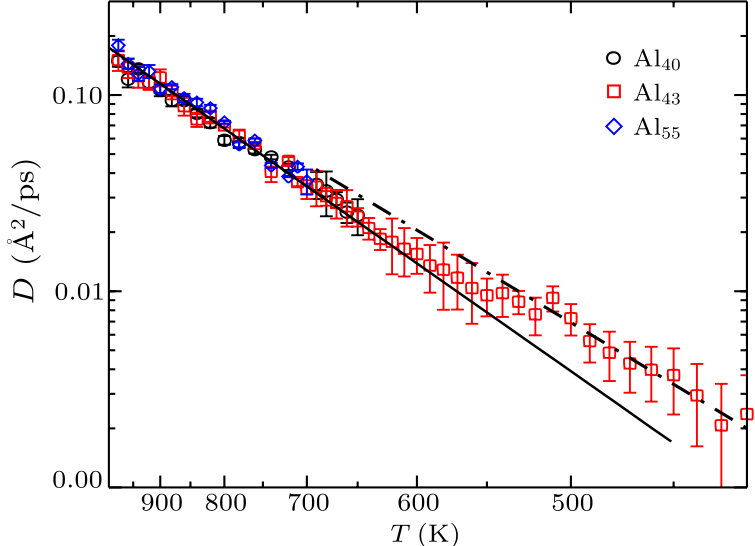
Fig. 2. (Color online) Diffusion constants versus temperature for Al$_{40}$, Al$_{43}$ and Al$_{55}$, where the $y$-axis is logarithmic and the $x$-axis is reciprocal temperature. The dashed and solid lines are the Arrhenius fits to the data. The difference in slope of the fitted lines indicates different activation energies.
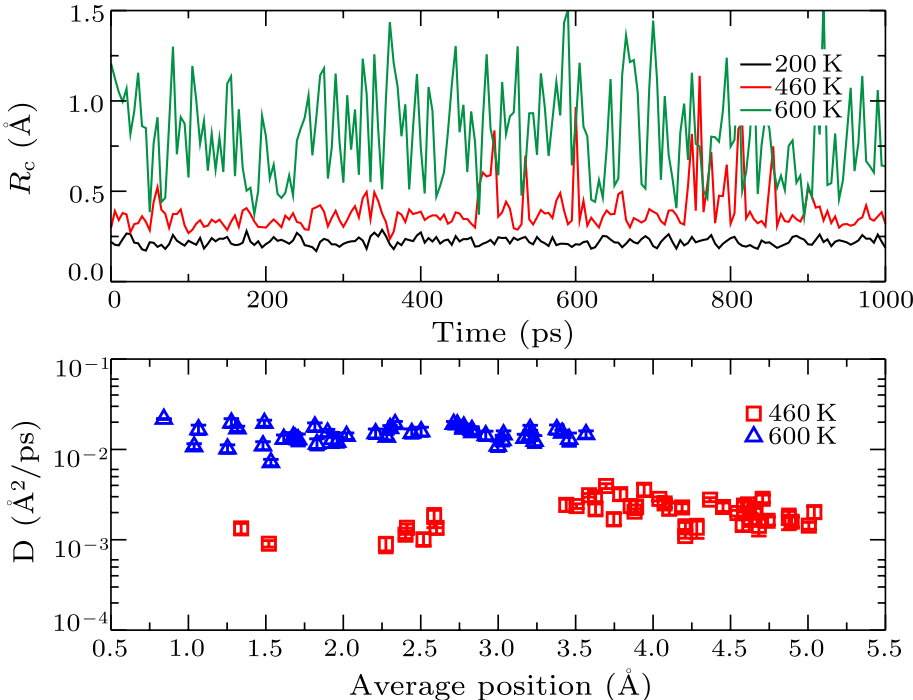
Fig. 3. (Color online) Upper panel: the time-dependent displacement for three temperatures. At 460 K, although the displacement is notable, it only occurs in several short durations, which indicates a cooperative motion of atoms in the glass transition range. Lower panel: the diffusion constant of each atom versus its average position. In the glass transition range, the inner atoms have non-negligible diffusion.
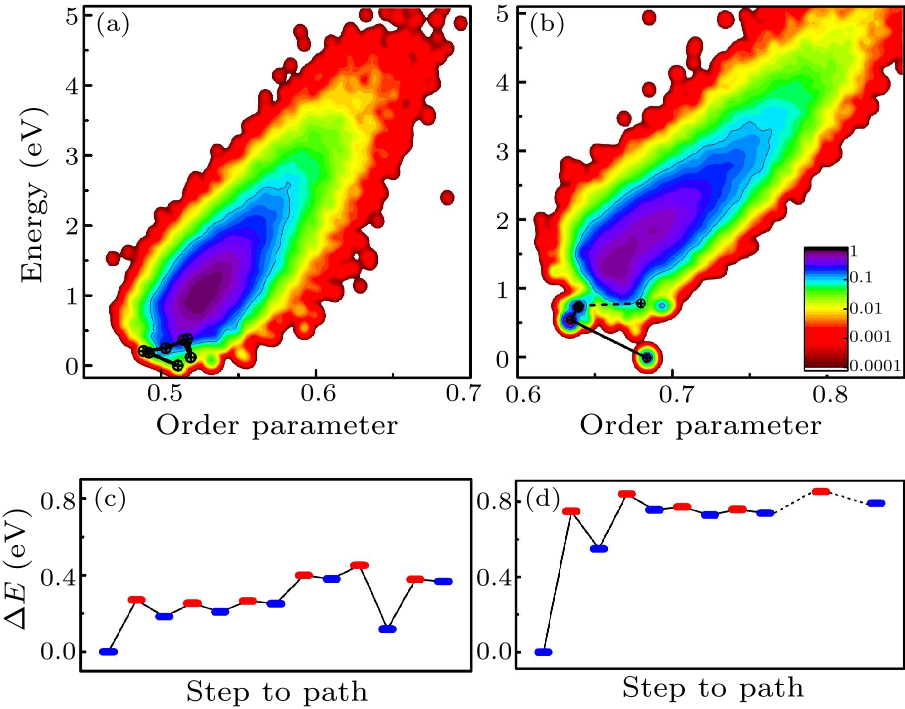
Fig. 4. (Color online) Upper panel: the phase diagram of Al$_{43}$ (left) and Al$_{55}$ (right), where the $x$ axis corresponds to the Steinhardt-type order parameter and the $y$ axis corresponds to the potential energy. The colors correspond to the visiting probability of minima during the SSW searches, with the highest probability after gauss broadening being scaled to 1. The black line shows the minima along a possible path for configuration changing from the ground state. Lower panel: the energy profile of the path indicated in upper panel (the black line). The blue bars correspond to the minima, while the orange bars correspond to the transition states. The energy of the global minimum is set to zero in both upper and lower panels.
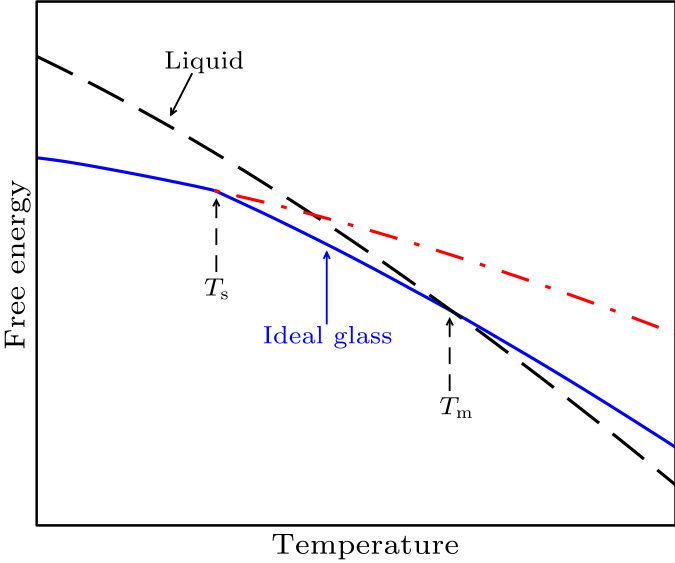
Fig. 5. (Color online) The schematic phase diagram for an ideal glass (blue solid line) and a liquid (black dashed line). $T_{\rm m}$ indicates the temperature at which the free energies of the liquid and ideal glass states are equal. $T_{\rm s}$ refers to the emergence of configuration entropies at this temperature. Without these configuration entropies, the free energy of an ideal glass should go along the red dot-dashed line for $T>T_{\rm s}$.
| [1] | Kennedy D and Norman C 2005 Science 309 75 | What Don't We Know?
| [2] | Angell C A 1995 Science 267 1924 | Formation of Glasses from Liquids and Biopolymers
| [3] | Debenedetti P G and Stillinger F H 2001 Nature 410 259 | Supercooled liquids and the glass transition
| [4] | Berthier L and Biroli G 2011 Rev. Mod. Phys. 83 587 | Theoretical perspective on the glass transition and amorphous materials
| [5] | Dyre J C 2006 Rev. Mod. Phys. 78 953 | Colloquium : The glass transition and elastic models of glass-forming liquids
| [6] | Cavagna A 2009 Phys. Rep. 476 51 | Supercooled liquids for pedestrians
| [7] | Wang W H 2012 Prog. Mater. Sci. 57 487 | The elastic properties, elastic models and elastic perspectives of metallic glasses
| [8] | Micoulaut M 2016 Rep. Prog. Phys. 79 066504 | Relaxation and physical aging in network glasses: a review
| [9] | Kauzmann W 1948 Chem. Rev. 43 219 | The Nature of the Glassy State and the Behavior of Liquids at Low Temperatures.
| [10] | Gibbs J H and DiMarzio E A 1958 J. Chem. Phys. 28 373 | Nature of the Glass Transition and the Glassy State
| [11] | Cohen M H and Turnbull D 1959 J. Chem. Phys. 31 1164 | Molecular Transport in Liquids and Glasses
| [12] | Angell C A 1968 J. Am. Ceram. Soc. 51 125 | Oxide Glasses in Light of the “Ideal Glass” Concept: II, Interpretations by Reference to Simeple Ionic Glass Behavior
| [13] | Kirkpatrick T R, Thirumalai D and Wolynes P G 1989 Phys. Rev. A 40 1045 | Scaling concepts for the dynamics of viscous liquids near an ideal glassy state
| [14] | Cammarota C and Biroli G 2012 Proc. Natl. Acad. Sci. USA 109 8850 | Ideal glass transitions by random pinning
| [15] | Sun D Y and Gong X G 1998 Phys. Rev. B 57 4730 | Structural properties and glass transition in clusters
| [16] | Noya E G, Doye J P K and Calvo F 2006 Phys. Rev. B 73 125407 | Theoretical study of the melting of aluminum clusters
| [17] | Neal C M, Starace A K and Jarrold M F 2007 J. Am. Soc. Mass Spectr. 18 74 | Ion calorimetry: Using mass spectrometry to measure melting points
| [18] | Shang C and Liu Z P 2013 J. Chem. Theory Comput. 9 1838 | Stochastic Surface Walking Method for Structure Prediction and Pathway Searching
| [19] | Zhang X J, Shang C and Liu Z P 2013 J. Chem. Theory Comput. 9 3252 | From Atoms to Fullerene: Stochastic Surface Walking Solution for Automated Structure Prediction of Complex Material
| [20] | Doye J P K 2003 J. Chem. Phys. 119 1136 | A model metal potential exhibiting polytetrahedral clusters
| [21] | Shang C, Zhao W N and Liu Z P 2015 J. Phys.: Condens. Matter 27 134203 | Searching for new TiO 2 crystal phases with better photoactivity
| [22] | Wei G F, Shang C and Liu Z P 2015 Phys. Chem. Chem. Phys. 17 2078 | Confined platinum nanoparticle in carbon nanotube: structure and oxidation
| [23] | Ercolessi F and Adams J B 1994 Europhys. Lett. 26 583 | Interatomic Potentials from First-Principles Calculations: The Force-Matching Method
| [24] | Sun D Y and Gong X G 2002 J. Phys.: Condens. Matter 14 L487 | A new constant-pressure molecular dynamics method for finite systems
| [25] | Steinhardt P J, Nelson D R and Ronchetti M 1983 Phys. Rev. B 28 784 | Bond-orientational order in liquids and glasses
| [26] | Zhang X J, Shang C and Liu Z P 2017 Phys. Chem. Chem. Phys. (accepted) | Pressure-induced Silica Quartz Amorphization Studied by Iterative Stochastic Surface Walking Reaction Sampling
| [27] | Adam G and Gibbs J H 1965 J. Chem. Phys. 43 139 | On the Temperature Dependence of Cooperative Relaxation Properties in Glass‐Forming Liquids
| [28] | Tombari E, Ferrari C, Salvetti G and Johari G P 2008 Phys. Rev. B 77 024304 | Vibrational and configurational parts of the specific heat at glass formation
| [29] | Goldstein M 1969 J. Chem. Phys. 51 3728 | Viscous Liquids and the Glass Transition: A Potential Energy Barrier Picture
| [30] | Grigera T S, Cavagna A, Giardina I and Parisi G 2002 Phys. Rev. Lett. 88 055502 | Geometric Approach to the Dynamic Glass Transition